3D genome - improving
Hi-C resolution

We developed two tools (HiCNN and HiCNN2) for resolution enhancement of Hi-C data. HiCNN uses a deep convolutional neural network (54 layers) to learning the mapping between low-resolution and high-resolution Hi-C contact matrices. HiCNN2 is an improved version of HiCNN. HiCNN2-enhanced high-resolution Hi-C data achieve smaller mean squared error and higher Pearson’s correlation coefficients with experimental high-resolution Hi-C data compared with existing methods HiCPlus and HiCNN. Moreover, HiCNN2 can recover more significant interactions detected by Fit-Hi-C compared to HiCPlus and HiCNN.
3D genome - normalizing
Hi-C and GAM data
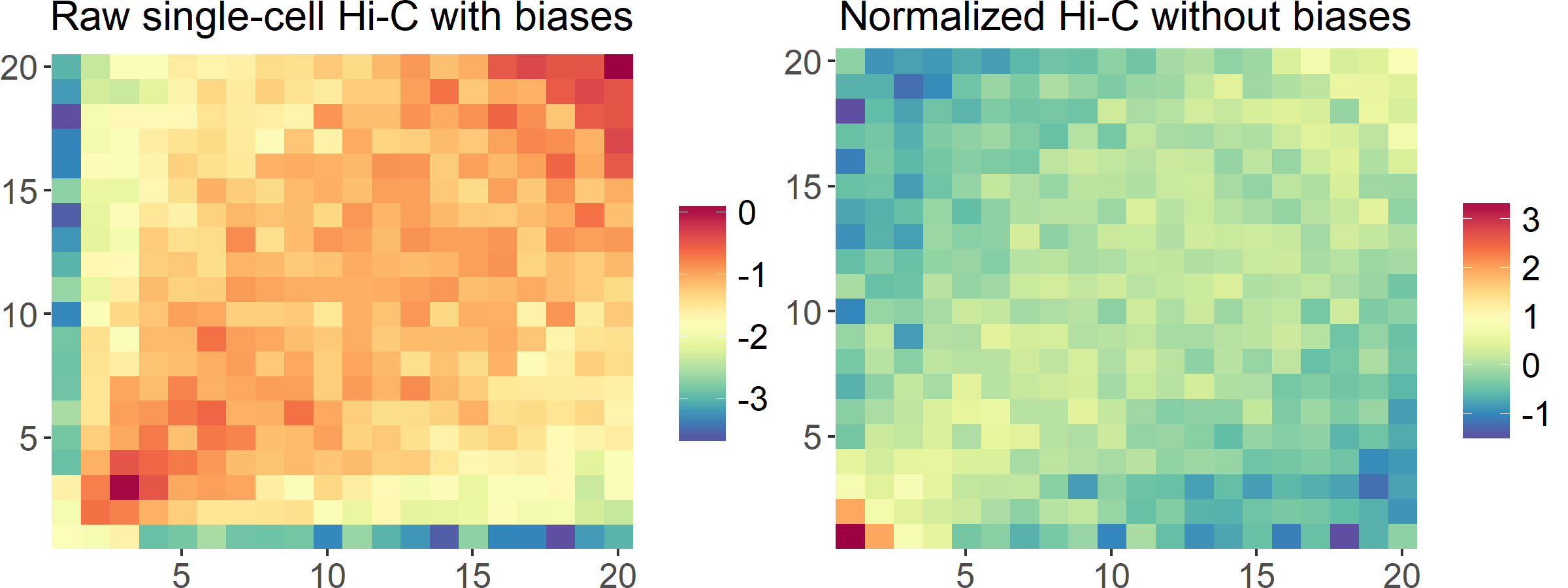
For normalizing single-cell Hi-C data, we developed scHiCNorm, which uses zero-inflated (Poisson and Negative Binomial) and hurdle (Poisson and Negative Binomial) models to remove biases, including cutting sites, GC content, and mappability. For normalizing GAM data, we developed an R package, which implements five normalization methods, including normalized linkage disequilibrium (NLD), vanilla coverage (VC), sequential component normalization (SCN), iterative correction and eigenvector decomposition (ICE), and Knight-Ruiz 2-norm (KR2).
3D genome - reconstructing
structures of chromatins and TADs
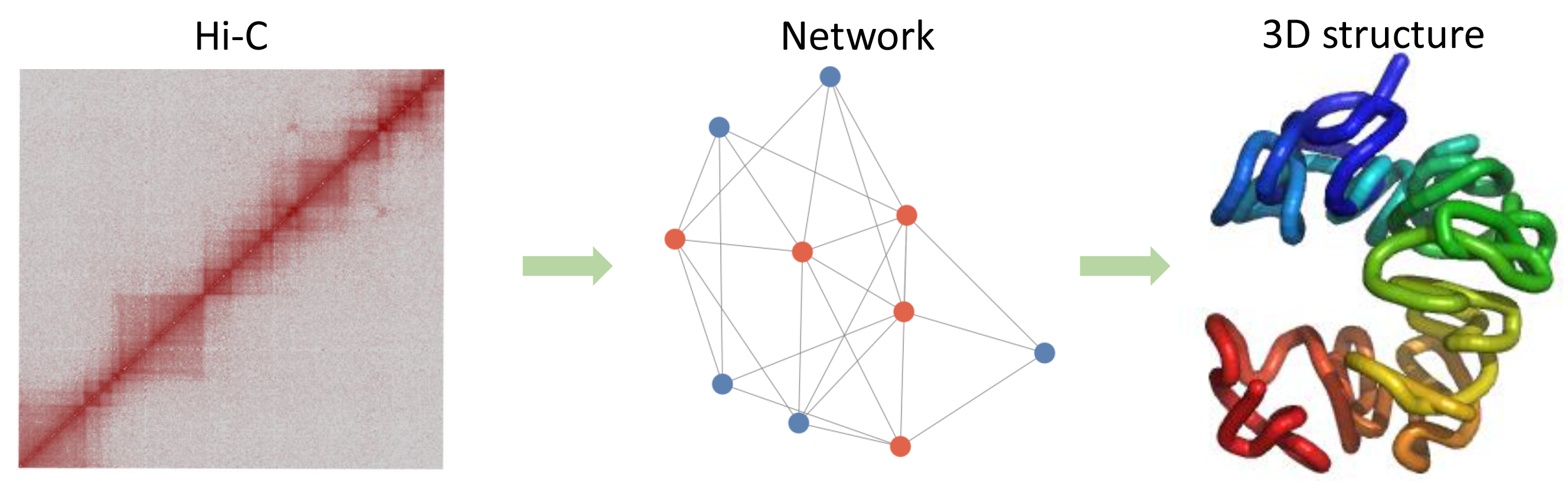
HiCNet is a tool capable of converting Hi-C contacts to spatial distances. And it can also infer three-dimensional structures using spatial distances and multidimensional scaling methods. TADKB is an integrated resource for exploring topologically associating domains (TADs). 3DChrom is designed to measure the three-dimensional structural properties of TADs.
Protein - SOV
Quality Assessment
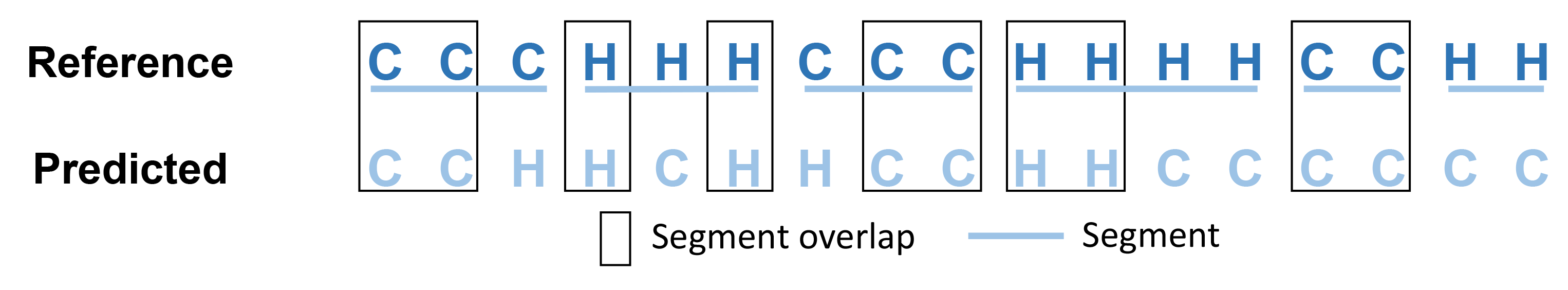
SOV_refine is a refined definition of segment overlap score based on the previous definitons of SOV scores, which is widely used to evaluate the quality of protein secondary structure predictions
MASS: Protein Single-Model Global Quality Assessment by Random Forest and Various Energies